Der Medizintechnikmarkt in den USA ist weltweit bei weitem der umsatzstärkste und wichtigste Einzelmarkt und wird es auch in Zukunft bleiben. Dennoch stellt sich die Frage, ob die Größe des Marktes allein ausschlaggebend für die First-Market-Entry-Entscheidung von Medtech-Start-ups ist oder ob andere Faktoren bei der Entscheidung eine Rolle spielen. Von Dr. Michael Thiel und Sonja Terszowski
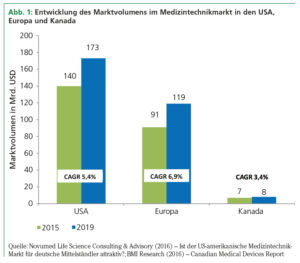
Der weltweite Medizintechnikmarkt wird auf 324 Mrd. USD im Jahre 2015 geschätzt, mehr als 70% davon machen Nordamerika und Europa aus. Größter Einzelmarkt sind die USA mit einem Umsatz in Höhe von 140 Mrd. USD im Jahre 2015, gefolgt von Europa mit 91 Mrd. USD. Der kanadische Medizintechnikmarkt beläuft sich auf 7 Mrd. USD (siehe Abb. 1). Der globale Medizintechnikmarkt ist nach wie vor ein großer Wachstumsmarkt – die CAGR bis zum Jahre 2019 wird für Europa mit 6,9%, für die USA mit 5,4% und für Kanada mit 3,4% prognostiziert. Wachstumstreiber sind die älter werdende Bevölkerung, der medizinisch-technische Fortschritt sowie der große Nachholbedarf an Medizinprodukten in den Emerging Markets.
Zulassung von Medizinprodukten
Medizinprodukte kommen im oder am Menschen zum Einsatz und bergen daher ein gewisses Risiko für den Patienten. Europa, Kanada und die USA haben daher spezielle Anforderungen und Richtlinien erlassen, um die Sicherheit und Funktionstüchtigkeit sowie die Qualität der Medizinprodukte sicherzustellen.

Auf dem europäischen Markt ist für den Handel mit Medizinprodukten eine CE-Kennzeichnung der Produkte erforderlich. Um diese Kennzeichnung zu erhalten, müssen Hersteller in einem Konformitätsbewertungsverfahren nachweisen, dass ihr Produkt mit den entsprechenden europäischen Medizinprodukterichtlinien konform ist. Medizinprodukte werden den Risikoklassen I, IIa, IIb oder III zugeordnet. Bei Produkten der Klasse I kann der KonKonformitätsnachweis in Eigenverantwortung des Herstellers erbracht werden. Für alle anderen Produktklassen muss eine benannte Stelle, beispielsweise der TÜV, in die Konformitätsbewertung eingebunden werden. Hersteller können dabei je nach Risikoklasse des Produktes zwischen unterschiedlichen Bewertungsverfahren wählen. Im Anschluss an die erfolgreiche Konformitätsbewertung muss der Hersteller in einer Konformitätserklärung versichern, dass sein Produkt die entsprechenden Richtlinien erfüllt.
In Kanada müssen alle Hersteller ihre Medizinprodukte bei der Gesundheitsbehörde Health Canada registrieren lassen. Health Canada unterscheidet zwischen Produkten der Klasse I, II, III und IV. Für Produkte der Klasse I ist lediglich ein Antrag für eine Medical Device Established License (MDEL) zu stellen. Bei Produkten der Klasse II bis IV müssen Hersteller eine Medical Device License (MDL) beantragen. Zudem müssen sie ein zertifiziertes Qualitätsmanagementsystem vorweisen, welches die kanadischen Medizinproduktevorschriften (Canadian Medical Device Regulations) erfüllt. Die Zertifizierung des Qualitätsmanagementsystems darf ausschließlich von anerkannten Canadian-Medical-Device-Assessment-System-Registraren vorgenommen werden.
Für den Handel mit Medizinprodukten in den USA ist eine Zulassung durch die FDA erforderlich. Diese unterteilt Medizinprodukte in Produkte der Klasse I, II und III. Hersteller von Produkten der Klasse I benötigen in der Regel keine Zulassung für ihre Produkte, müssen jedoch die Registrierungsanforderungen der FDA erfüllen. Bei vergleichbaren Produkten der Klasse II, in einigen Fällen auch der Klasse I oder III, die in den USA bereits auf dem Markt vertrieben werden oder wurden, ist ein Antrag auf eine Premarket Notification – auch 510(k)-Antrag – ausreichend. Mit höherem Risiko behaftete Produkte der Klasse III müssen überwiegend das Premarket-Approval (PMA)-Verfahren durchlaufen. Hierbei muss der Hersteller im Vorfeld eine Investigational Device Exemption (IDE) beantragen, um alle geforderten Studien durchführen zu können. Neuartige Produkte, die nicht mit bereits klassifizierten Produkten vergleichbar sind, werden automatisch der Klasse III zugeordnet. Bei Produkten mit geringerem Risiko kann ein Antrag auf eine De-Novo-Klassifizierung gestellt werden, damit diese Produkte in Produkte der Klasse I oder II umklassifiziert werden. Alle Hersteller müssen unabhängig von der Klassifizierung ihre Betriebsstätte bei der FDA registrieren lassen und die zu vermarktenden Produkte in der Datenbank der FDA listen. Zudem muss bei Herstellern ohne Niederlassung in den USA ein in den USA ansässiger Ansprechpartner – US Search Agent – benannt werden.

Präferenzen bei der Wahl des First-Market-Entry für Medtech-Start-ups
Wird unter Einbeziehung der im letzten Abschnitt dargestellten Genehmigungsprozesse für Medizinprodukte nach der präferierten Region für den First-Market-Entry für Medtech-Start-ups gefragt, zeigen unsere Erfahrungen, dass vorwiegend Europa als präferierte Region für den First-Market-Entry gewählt wird. Der Grund liegt darin, dass in Europa, dem zweitgrößten globalen Medizintechnikmarkt, die Markteinführung und damit der „Proof-of-Marketability“ für ein Produkt wesentlich schneller erreicht werden kann als in den USA, da die CE-Zertifizierung für das Produkt schneller und mit weniger Aufwand zu bekommen ist. In Kanada und den USA werden die Medizinprodukte durch staatliche Behörden zugelassen, was zeitaufwendig und kostspielig ist. In der EU gibt es dagegen keine Zulassung im klassischen Sinn. Zudem fordert die FDA oft einen Nachweis der Sicherheit und Wirksamkeit der Produkte durch aufwendige klinische Studien. In Europa muss die Eignung für den vorgesehenen Verwendungszweck nachgewiesen werden. Weiterhin erhalten Produkte durch die CE-Zertifizierung einen „europäischen Reisepass“1 und können im gesamten europäischen Wirtschaftsraum vertrieben werden. In den USA bestehen auch deutlich höhere Haftungsrisiken, die teilweise mit hohen Schadensersatzforderungen
bei Produkthaftungsfällen verbunden sind. Ein weiterer Aspekt, der nicht vernachlässigt werden darf, ist die Transparenz der Daten. Die FDA veröffentlicht alle eingereichten Daten zur Sicherheit und Wirksamkeit im Internet, in Europa sind diese nicht öffentlich und somit dem Wettbewerb nicht zugänglich.
Fazit
Die USA werden ihre Stellung als größter Medizintechnikmarkt in den kommenden Jahren weiter ausbauen. Dennoch entscheiden sich viele nordamerikanische Medtech-Start-ups dafür, ihr Produkt zuerst auf dem europäischen Markt einzuführen. Dies liegt vor allem an dem einfacheren und schnelleren CE-Zertifizierungsprozess für Medizinprodukte in Europa – womit der „Proof-of-Marketability“ des Produktes auch schneller gezeigt
werden kann.
Autor/Autorin
Die GoingPublic Redaktion informiert über alle Börsengänge, Being Public, Investor Relations, Tax & Legal, Themen und Trends rund um die Hauptversammlung sowie Technologie – Finanzierung – Investment in den Lebenswissenschaften.